معرفی بیماری موکوپلی ساکاریدوز یا سندرم هارلر
بیماری موکوپلی ساکاریدوز ها (MPS) شامل گروهی از بیماری های متابولیک با ریشه ژنتیکی می باشند. این بیماری که با نام دیگر سندرم هارلر که یک بیماری نادر ژنتیکی است و در اثر جهش در ژن آنزیمهای مربوط به تجزیه در موکوپلی ساکاریدها، به وجود میآید. عملکرد نادرست آنزیمهای لیزوزومی خاص منجر به تجمع غیرطبیعی برخی از کربوهیدراتهای پیچیده در شریان ها و اسکلتهای بدن میشود. تجمعاتی که ممکن است در سیستم تنفسی، کبد، طحال، سیستم عصبی مرکزی، خون و مغز استخوان نیز دیده شوند. این تجمع در نهایت باعث آسیب تدریجی به سلولها، بافتها و سیستمهای مختلف اندام بدن می شود
براساس تفاوت های بالینی و ژنتیکی، شش موکوپلی ساکاریدوز متفاوت شناسایی شده است. بررسی بیوشیمیایی توسط دانشمندان نشان داده است که انواع گوناگون از کمبود آنزیم های منفرد متفاوتی ناشی می شوند و همه آنها به استثنای نشانگان هانتر که وابسته به X است، به شکل مغلوب اتوزومی به ارث میرسند. انواع مختلف این بیماری به شرح زیر است :

سندرم هانتر (MPS-II) :
بیماران مبتلا به سندرم هانتر معمولا در هنگام تولد علائم بالینی را نشان میدهند و در حوالی دو تا چهار سالگی علائم بسیار مشخص تر میشود. علائمی مثل دست دادن شنوایی، یک تاریخچه بازگشت عفونت، اسهال و رشد ضعیف که در سنین ۲ تا ۵ سالگی ظاهر میشود. کودکان مبتلا همچنین ممکن است گردن کوتاه، سر بزرگ غیرطبیعی، قفسه سینه پهن، تاخیر در رویش دندان، کاهش شنوایی پیشرونده، داشته باشند. علاوه بر این موارد، پیشرفت آهستهتر بیماری در افرادی که شکل خفیف این اختلال را دارند رخ میدهد. درمان موکوپلی ساکاریدوز نوع هانتر متشکل از دو جزء، مدیریت علایم و جایگزینی کمبود آنزیم با درمان جایگزینی آنزیم (ERT) است.
سندرم هورلر (MPS-I) :
شدید ترین نوع موکوپلی ساکاریدوز است. افراد مبتلا در سالهای اول زندگی، کودکان خمیدگی بخش انتهایی نخاع و کدورت قرنیه شده و در سال دوم دچار سختی مهره ها، چهره خشن و کبد و طحال بزرگ شده میشوند. همچنین رشد ذهنی در حدود دو سالگی شروع به پسرفت می کند. پیشرفت این علائم همراه با زوال عقلی و سرانجام به فوت در اواسط نوجوانی، به دلیل ترکیبی از نارسایی قلبی و عفونتهای ریوی پیش می آید. متاسفانه هنوز درمانی برای بیماری موکوپلی ساکاریدوز نوع MPS I وجود ندارد بنابراین درمان بر تسکین علائم متمرکز شده است.
سندرم مورکیو (MPS-IV) :
افراد مبتلا ممکن است علائمی مثل گردن کوتاه غیرعادی، عقب ماندگی رشد، برجسته بودن پایین صورت، صافی کف پا، زانوهایی که به طور غیر طبیعی نزدیک به هم هستند، غیر طبیعی بودن پهلوها و… است. در این افراد سطح هوشیاری طبیعی و طول عمر طولانی مدت است، اگرچه خطر به هم فشردگی طناب نخاع ناشی از پیشرفت درگیری اسکلتی وجود دارد. برای درمان بیماری موکوپلی ساکاریدوز نوع مورکیو میتوان از ژن درمانی استفاده کرد.
سندرم سان فیلیپو (MPS-III) :
علائم اولیه سندرم سان فیلیپو از زیر شاخه های موکوپلی ساکاریدوز شامل اختلالات خواب، بیش فعالی و تاخیر در یادگیریه راه رفتن، سینه خیز رفتن است. این افراد در سنین اولیه بزرگسالی به سمت تشنج و حتی مرگ پیش میروند. همچنین ممکن است تشنج، راه رفتن ناپایدار و رفتار پرخاشگرانه را تجربه کنند. در حال حاظر هیچ درمانی برای بیماری سان فلیپو وجود ندارد. در اغلب موارد، درمان به کاهش این اختلال توسط متخصصین مغز، چشم پزشکان، متخصصین قلب، دنداپزشکان و مشاوران ژنتیک محدود شده است.
سندرم ماراتولامی (MPS-VI) :
موکوپلی ساکاریدوز نوع ماراتولامی باعث میشود بدن در تجزیه مولکولهای بزرگ قند در سلول ها مشکل داشته باشد. این باعث التهاب و زخم شدن تعدادی از بافت ها و اندام های بدن میشود. کودکان مبتلا با ویژگی های شبیه نشانگان هورلر مشتمل بر قد کوتاه یا بدشکلی سینهای، بزرگ شدن غیر طبیعی کبد، صورت زبر، انقباضات مفصلی، و محدودیت حرکت مفصلی ظاهر مییابند و در بیشتر موارد، هوش طبیعی است. برای درمان نوع ماراتولامی این بیماری ( سندرم هارلر ) میتوان عمل جراحی انجام داد ولی قبل از عمل جراحی از آنجایی که کودکان مبتلا به سندرم مارفان نیز ممکن است مشکلات قلبی و ریوی داشته باشند، برای درمان اسکولیوز قبل از عمل جراحی، مشاوره و معاینه با متخصص قلب و ریه ضروری است.
(بیماری MPS VII) سندرم اسلای
این بیماری یکی از نادرترین انواع بیماری موکوپلی ساکاریدوز است و حدودا از هر ۲۵۰,۰۰۰ نوزاد ۱ نفر را تحت تاثیر قرار می دهد. در موارد خیلی نادر، MPS VII سبب می شود که کودکان با خیز جنینی یا هیدروپس فتالیس متولد شوند. در هیدروپس فتالیس مقدار زیادی از مایعات در بدن جمع می شود. رهایی از این مشکل معمولا چندین ماه یا کمتر زمان می برد. بیشتر کودکانی که دچار MPS VII هستند معمولا خیلی تحت تاثیر بیماری قرار نمی گیرند. علائم نورولوژیکی این بیماری شامل این مواردند: اختلالات ذهنی خفیف تا متوسط تا ۳ سالگی، هیدروسفالی ارتباطی، تحت فشار بودن عصب ها، تیره و تار شدن قرنیه و از دست دادن دیدن شب.
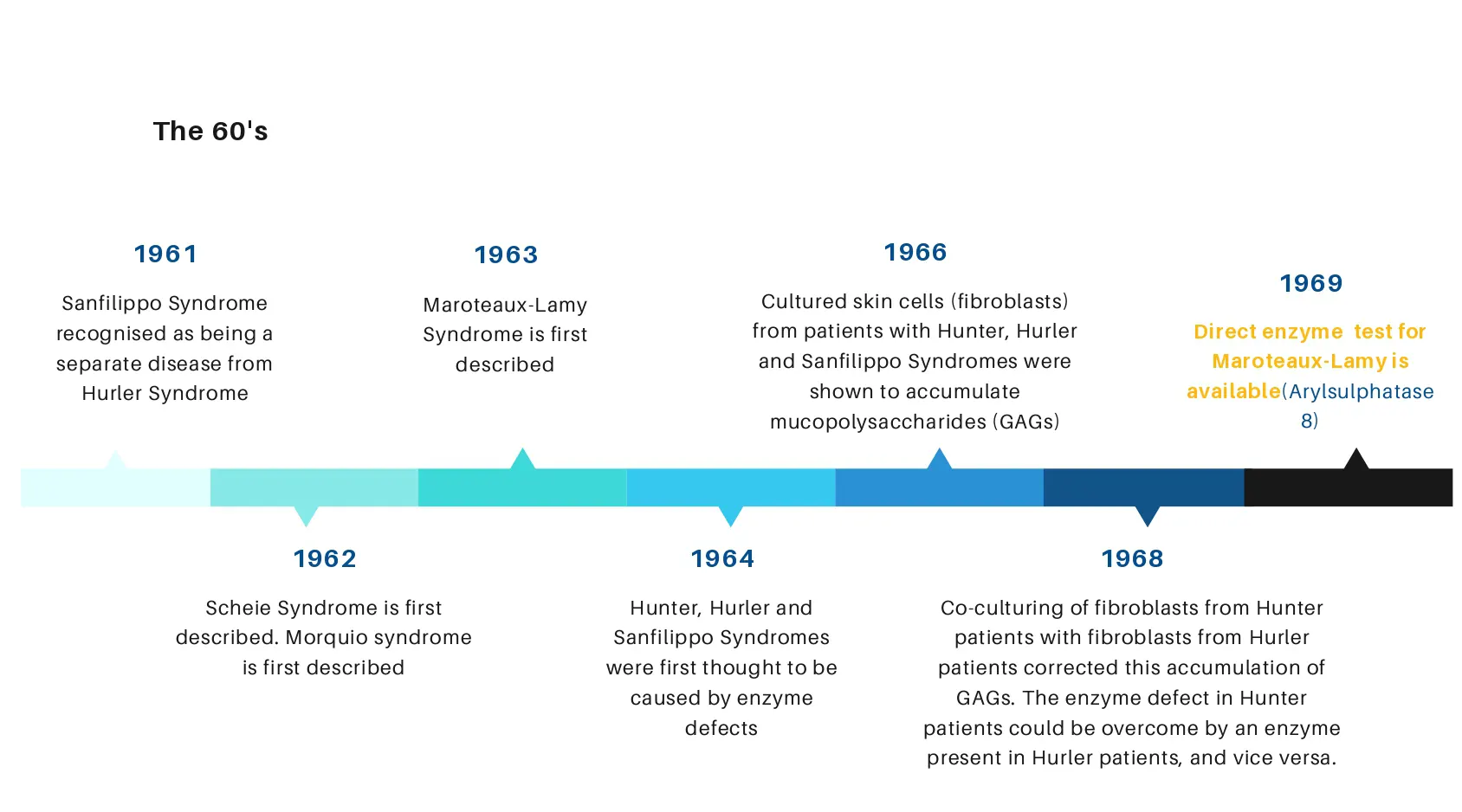
تاریخچه بیماری موکوپلی ساکاریدوز
برای اینکه متوجه شوید بیماری موکوپلی ساکاریدوز چیست ناچارید از تاریخچه این بیماری اطلاع داشته باشید.
این اختلال (سندرم هارلر یا ساکاریدوز) از اوایل قرن ۲۰ آغاز شد، در زمانی که کودکی به مرکز درمانی ادینبورگ برده شد؛ این بیمار تمام ویژگی های بیماری ای که بعدها به بیماری موکوپلی ساکاریدوز شناخته شد را داشت. بخاطر این که در آن زمان چیزی درباره این بیماری نمی دانستند، پزشک معالج وی به طور موقت نام این بیماری را Johnny McL’s disease گذاشت. بعد ها در سال ۱۹۱۷ دو مورد از این بیماری در شهر وینیپگ گزارش شد.
دو سال بعد، یک مورد از این بیماری در کشور آلمان گزارش شد. تمام این بیماران تقریبا علائم مشابهی داشتند. همه آن ها نشانه هایی شامل : کبد و طحال بزرگ شده، پنجه ای شدن دست ها و بدشکلی های استخوانی را داشتند. بیماری فرد آلمانی به شدت پیشرفت کرده بود و او در مراجعه به پزشک تعلل کرده و برای رجوع به پزشک بسیار تاخیر کرده بود.
گزارشات و دست نوشته های پزشکان معالج این بیماران بعدها توسط سایر پزشکان ژنتیک مطالعه شد و نشان داد که کودکان دیگری هم بودند که مانند این بیماران بودند و خصوصیات یکسانی داشتند. بعد از آن نامی که برای این اختلال یا بیماری انتخاب کرده بودند، سندرم هارلر بود.همچنین باید خاطر نشان کرد که توصیف و مشاهده اولیه این بیماری در کشور کانادا صورت گرفته بود.
بررسی این بیماری و مشاوره در خصوص بارداری و انجام آزمایشات ژنتیکی جهت تشخیص و درمان این بیماری در آزمایشگاه ژنتیک ژن آزما توسط کادر مجرب و حرفه ای انجام میپذیرد ؛ جهت دریافت نوبت مشاوره حضوری یا آنلاین با ما تماس بگیرید.
بدون دیدگاه