
تشخیص ژنتیکی آتروفی های عضلانی نخاعی
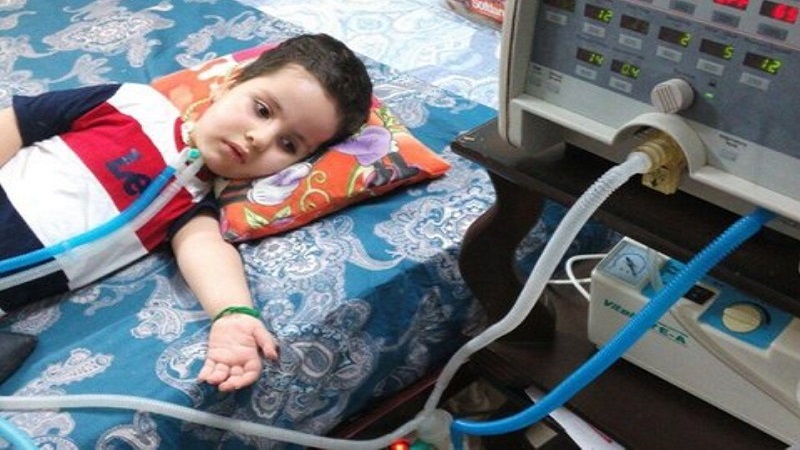
تشخیص ژنتیکی آتروفی های عضلانی نخاعی : تشخیص ژنتیکی مولکولی به وسیله پروب DNA در نمونه خون یا بیوپسی عضله یا پرزهای کوریونیک، برای تشخیص در موارد مشکوک و نیز تشخیص قبل از تولد در دسترس می باشد. اکثر موارد بیماری به صورت اتوزوم مغلوب به ارث
می رسند. میزان بروز SMA، ۱۰-۱۵ مورد از هر ۰۰۰, ۱۰۰ تولد زنده بوده و تمام گروه های نژادی را درگیر می کند. این بیماری دومین بیماری نوروماسکولار شایع بعد از دیستروفی عضلانی دوشن می باشد. میزان بروز هتروزیگوتی در SMA اتوزومال مغلوب 50: 1 می باشد.
لوکوس ژنتیکی | تشخیص ژنتیکی آتروفی های عضلانی نخاعی
برای هر سه فرم شایع SMA روی کروموزوم ۵ و حذفی در لوکوس 13q-q11 5می باشد و نشان می دهد که آنها بیش از آنکه بیماریهای مختلفی با واریانت های یک بیماری هستند. ژن درگیر SMINT وزن مولکولی KDa ۳۸داشته و دارای ۸ اگزون می باشد که ۲۰ کیلوباز بوده و دارای اگزون های تلومریک و سنترومریک می باشد که فقط bp 5با هم تفاوت داشته و یک نسخه دارای ۲۹۴ اسید آمینه راکد میکنند. ژن SMN1 در یک نسخه ژنی کاملا مشابه که SMN2 نامیده می شود تکثیر می شود و از روی هر دو ژن نسخه برداری صورت می پذیرد.
SMN2 در تمام بیماران مبتلا به SMA وجود دارد،
ولی نمی تواند به طور کامل نقص SMN1 را جبران نماید. از آنجایی که SMN2 توانایی کد کردن تعداد کمی پروتئین مشابه را دارد، ارتباط بین تعداد کپی SMN2 و شدت بالینی SMA توجیه می شود. تفاوت مهم بین ژن SMNI و SMN2 عبارت است از انتقال سیتوزین (C) به تیمین (T) در اگزون V ژن .SMN2 ژن دیگر، ژن مهارکننده آپوپتوز نورونی (NAIP)، درمجاورت ژن SMN قرار داشته و در بسیاری از موارد، دوبلیکاسیون معکوس با دو کپی، تلومریک و سانترومریک از هر دو ژن وجود دارد.
موتاسیون های مجزا یا حذفهای NAIP | تشخیص ژنتیکی آتروفی های عضلانی نخاعی
علائم بالینی SMA را ایجاد نکرده و یک ایزوفرم بدون عملکرد را تولید میکنند که فاقد آمینواسیدهای کربوکسی – ترمینوس کدشده توسط اگزون ۷ می باشد. فرمهای خفیفتر SMA، بیش از دو کپی از SMN2 دارند، و در بیماران دارای حذف هموزیگوس در ژن SMN1 که مبتلا به بیماری با شروع دیررس هستند، ۴ کپی از SMN2 وجود دارد. ژن دیگری در محل 13-21 q وq11 در SMA ممکن است توجیه کننده نارسایی تنفسی زودرس در بعضی بیماران باشد. گسترش نوکلئوتید (۳) فقط در ۱۰-۵٪ موارد SMA دیده میشود و مکانیسم ژنتیکی بیماری در اکثریت بیماران حذفها یا splicing out در اگزون ۷ و ۸ می باشد.
یک جفت ژن دیگر در مجاورت ژن های SMN1 و SMN2 به نامهای SERF1 و SERF2 نیز ممکن است. نقش ثانویه ای داشته باشند. پروتئین های سنتز شده توسل SMN، رشد أكسونال و الوكاليزاسيون بتااكتین در mRNA را در مخروط رشدی (cone) نورونهای حرکتی تنظیم می نماید. موارد کمی از خانواده های با توارث اتوزوم غالب مشخص شده اند و یک فرم نادر وابسته به X مغلوب هم وجود دارد. بررسی موارد ناقل به کمک dose analysis در دسترس می باشد.
آزمایشگاه ژنتیک پزشکی ژن آزما
آزمایشگاه ژنتیک اصفهان
آدرس:
اصفهان خیابان شریعتی غربی پلاک 208
ساعات کاری:
شنبه تا پنجشنبه 8 صبح الی ۱۱ شب
تلفن تماس:
۳۶۲۶۹۵۸۶-۷
geneazmalab.com
جهت مشاهده پیج اینستاگرام مرکز تخصصی ژنتیک پزشکی ژن آزما کلیک کنید.
مطالب بیشتر:
اختلال ژنتیکی نوروفیبروماتوز و علت آن
انواع دیستروفی میوتونیک و تفاوت آن
نکاتی در خصوص برنامه غذایی بیماران PKU

daktilogibigibi.nppYS4Ucrwwq